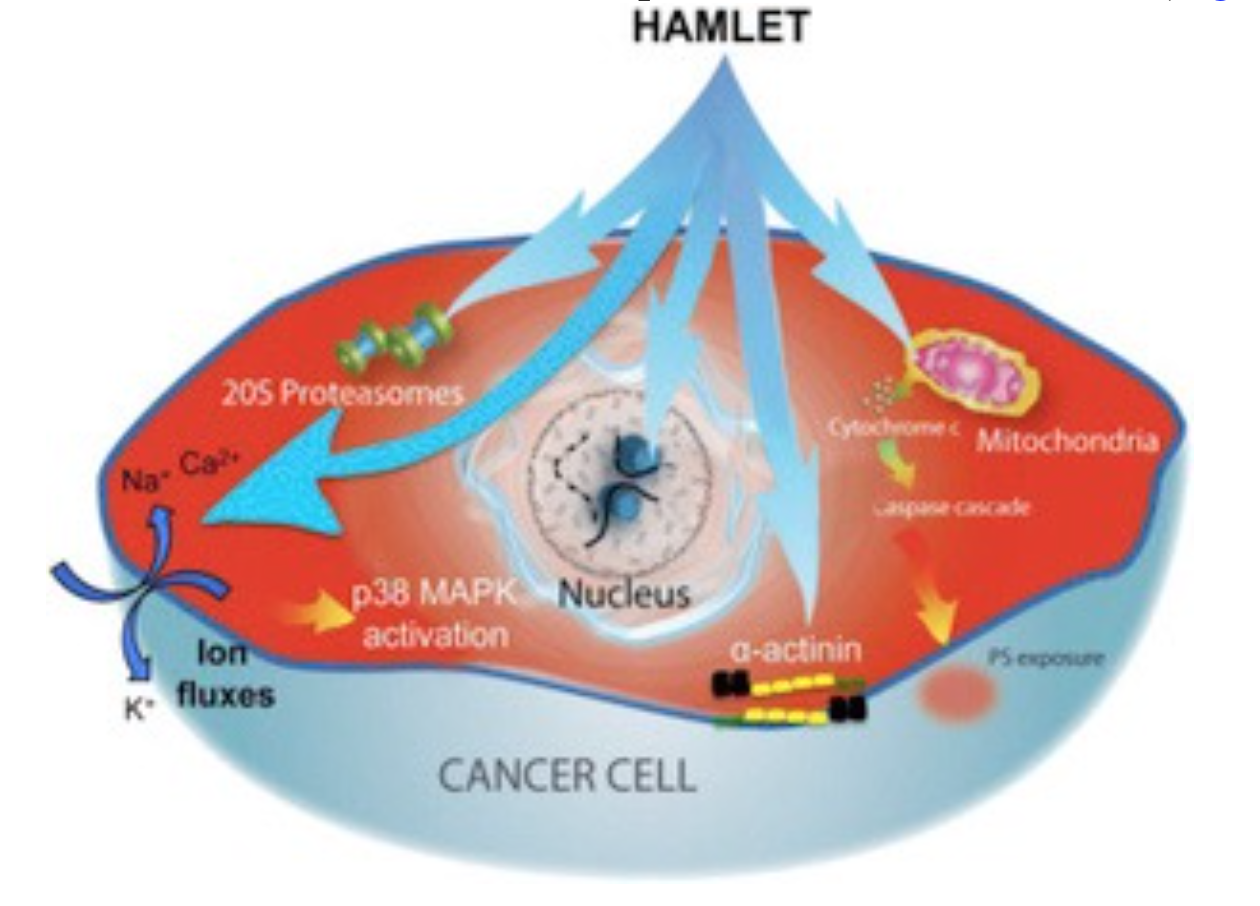
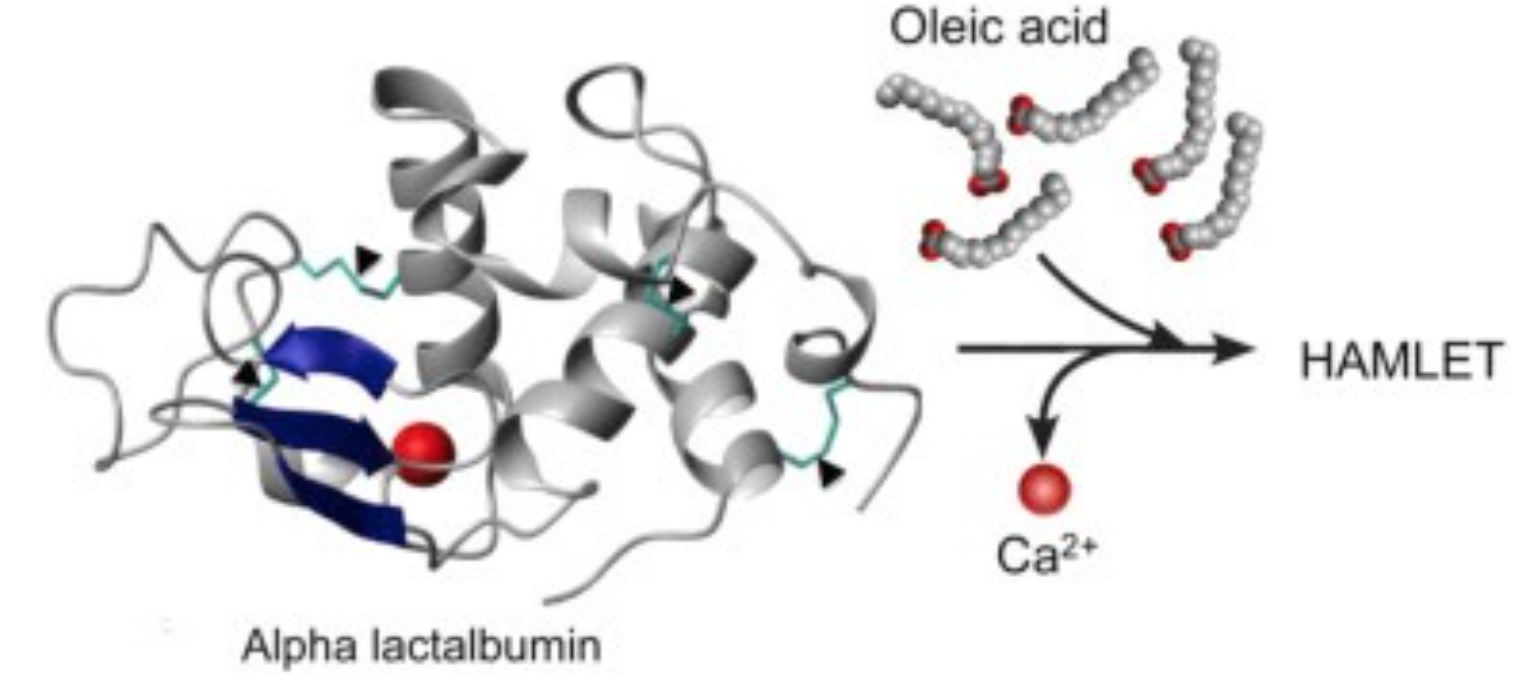
Cellular and molecular targets
2.2. Membrane perturbations and ion fluxes
HAMLET integrates into the membranes of tumor cells and triggers rapid ion fluxes, which activate downstream signaling pathways involved in cell death. Three critical features of the membrane response to HAMLET have been identified. (1) HAMLET interacts with lipid membranes in a protein receptor independent manner, as shown by the insertion of HAMLET into the membranes of giant unilamellar vesicles, disruption of the spherical shape and rapid tubulation, leading to a reduction in lumen size. (2) HAMLET creates a new membrane compartment for interactions with cellular targets and inhibits critical Ras oncogene driven signaling pathways. (3) This response to HAMLET does not occur in primary non-transformed cells, indicating tumor selectivity. The results suggest that HAMLET produces membrane conformations that serve as surrogate receptors for subsequent signal transduction, leading to tumor cell death. These effects are specific to the HAMLET complex, as the individual components of the complex, native alpha lactalbumin and oleic acid, did not affect the integrity of model membranes. The results indicate that both the partially unfolded protein and oleic acid are required to alter the structure of tumor cell membranes.
Ion fluxes regulate cellular homeostasis, through a multitude of effector functions. In parallel with the membrane changes, HAMLET activates rapid ion fluxes, with an influx of Na+ and Ca2+ and an efflux of K+ ions, as shown by real time confocal imaging, patch-clamping and fluorometry. The Na+ and K+ fluxes are thought to drive the cell death response, as ion flux inhibitors prevent HAMLET internalization, transcriptional responses and tumor cell death. These include amiloride, which inhibits the Na+/H+exchange and barium chloride (BaCl2), which inhibits K+ fluxes. HAMLET has not been shown to activate preformed ion channels, suggesting that channel-independent membrane permeabilization mechanisms might be involved.
Further mechanistic insights into this process have been gained, using synthetic alpha-helical peptides. Peptides covering the alpha1 and alpha2 domains of the protein triggered rapid ion fluxes in the presence of oleate and were internalized by tumor cells, causing rapid and sustained changes in cell morphology. The alpha peptide-oleate bound forms also triggered tumor cell death with comparable efficiency as HAMLET. In addition, shorter peptides corresponding to those domains were biologically active.
The results identify HAMLET as a membrane-perturbing agonist that triggers lethal ion fluxes in tumor cells.
2.3. Effects on nucleotide-binding proteins – kinases and GTPases
The ion fluxes activate a rapid p38 MAPK response as shown by transcriptomic analysis of HAMLET-treated tumor cells. In parallel, ERK1/2 phosphorylation is inhibited, consistent with the shift from proliferation to cell death. The activation of p38 and inhibition of ERK1/2 phosphorylation was reversed by ion flux inhibitors (amiloride or BaCl2), confirming the ion flux dependence. Importantly, pharmacological inhibitors of p38α and p38β delayed tumor cell death, as did p38-specific siRNAs. In contrast to tumor cells, normal differentiated cells showed weaker ion flux responses and less prominent changes in global transcription.
These findings suggested that affinity for conserved molecular motifs explains the apparent multitude of HAMLET targets in tumor cells. To identify such targets, we performed a proteomic screen of 8000 human proteins. By direct binding, we identified a large number of nucleotide binding proteins as HAMLET targets. These included 3 ATPases, 24 members of the Ras family of GTPases and 111 Kinases representing all branches of the kinome tree. In a kinase activity array, HAMLET acted as a pan-kinase inhibitor, reducing the activity of about 69% of the 476 kinases tested. This broad kinase inhibition was confirmed in protein lysates from HAMLET treated lung carcinoma cells, using an antibody microarray detecting phosphorylated proteins. Furthermore HAMLET was shown to co-localize with the Ras family of GTPases in membrane clusters and Ras activity was inhibited. The results identify HAMLET as an inhibitor of kinases and GTPases, to which tumor cells are addicted.
The results identify HAMLET as a potent and broad kinase inhibitor with specificity for tumor cells.
2.4. Chromatin interactions
In early studies our group has shown that HAMLET crosses the cytoplasmic membrane and accumulates in tumor cell nuclei. The accumulation of HAMLET in tumor cell nuclei was initially demonstrated by confocal microscopy of biotinylated HAMLET and fluorophore-labeled streptavidin and was confirmed using radiolabeled HAMLET. By far western blot and mass spectrometry analysis (MALDI-TOF), histones H2B, H3 or H4, were identified as nuclear HAMLET targets and HAMLET was shown to interfere with the formation of nucleosomes in the salt-jump assay. Furthermore, the sensitivity of tumor cells to HAMET is affected by chromatin acetylation, as histone deacetylase inhibitors open up the chromatin to HAMLET and synergistically kill tumor cells.
The results suggest that HAMLET may ‘seal the fate’ of dying tumor cells, through high affinity histone interactions and perturbation of the chromatin structure.
2.5. Proteasome inhibition
Proteasomes are crucial for extra-lysosomal protein degradation of endogenous misfolded proteins taking place in the barrel shaped 20S proteasome core. In a proteomic screen, catalytic proteasome subunits were identified as HAMLET targets. Furthermore, as α-lactalbumin is partially unfolded in the HAMLET complex, we hypothesized that the complex is targeted to the 20S proteasome for degradation. We found that HAMLET co-localizes with proteasomes in tumor cell cytoplasm and nuclei and detected an inhibitory effect of proteasome activity in whole cell extracts from HAMLET treated tumor cells. Interestingly, we also obtained structural evidence for proteasome disintegration by HAMLET, after incubation of HAMLET with intact 20S proteasomes, in vitro. Based on the above observations, we concluded that inhibition of proteasome activity might contribute to HAMLET-induced tumor cell death.
3. Structure of HAMLET complex