Newsletter
Read more >>

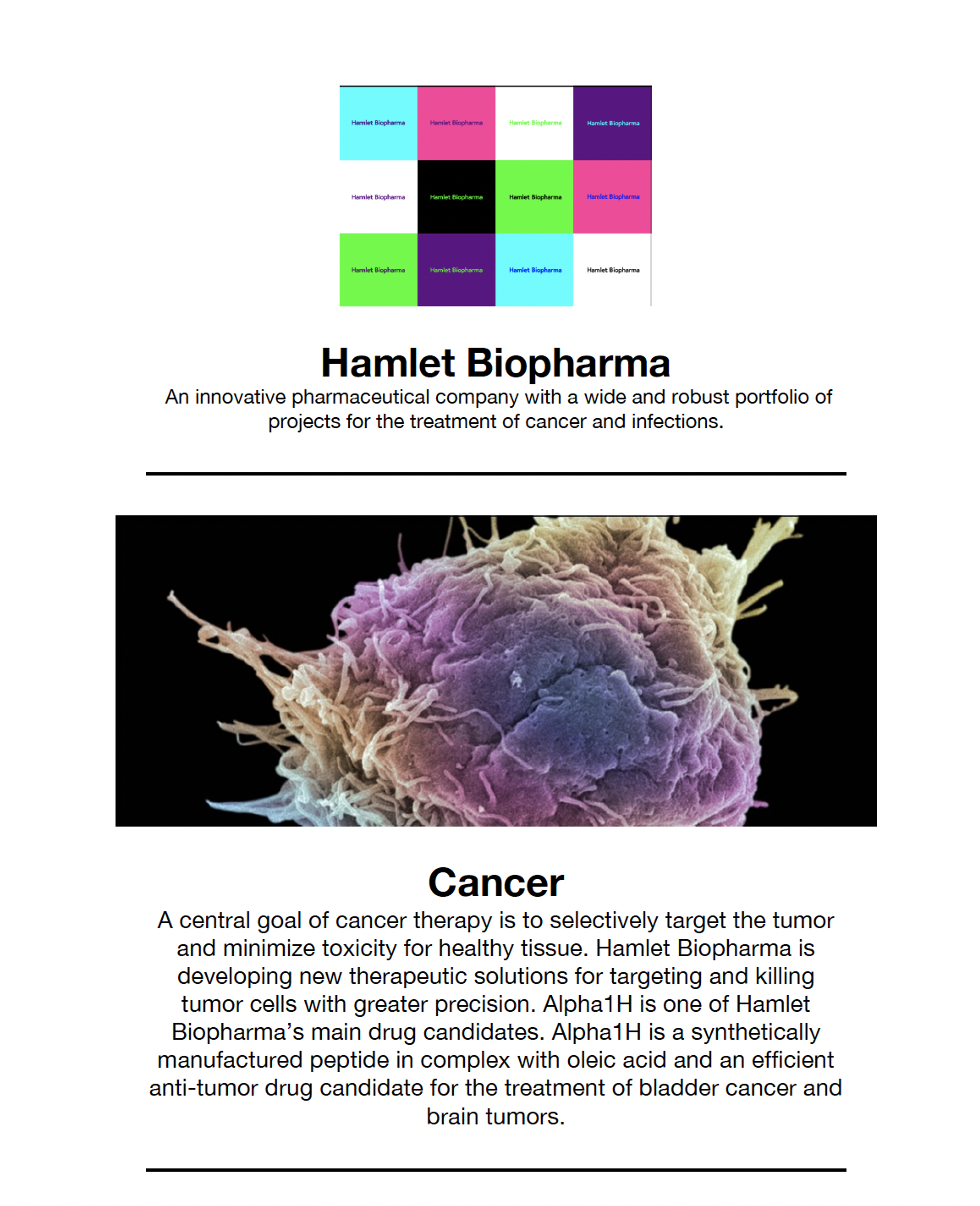
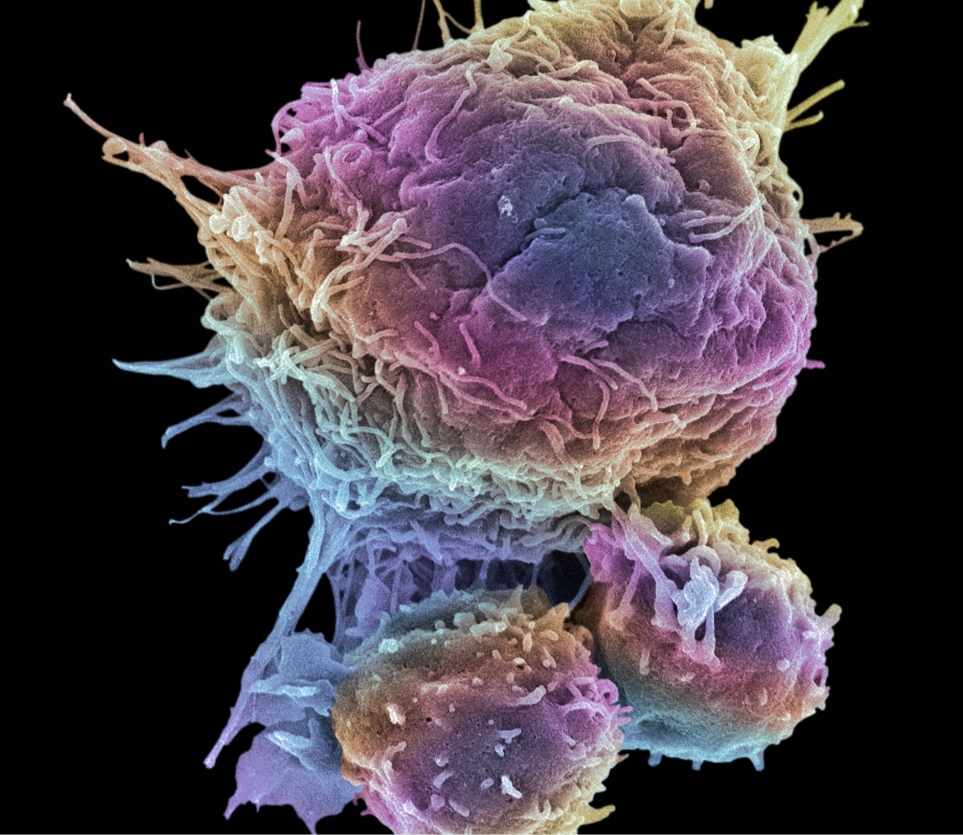
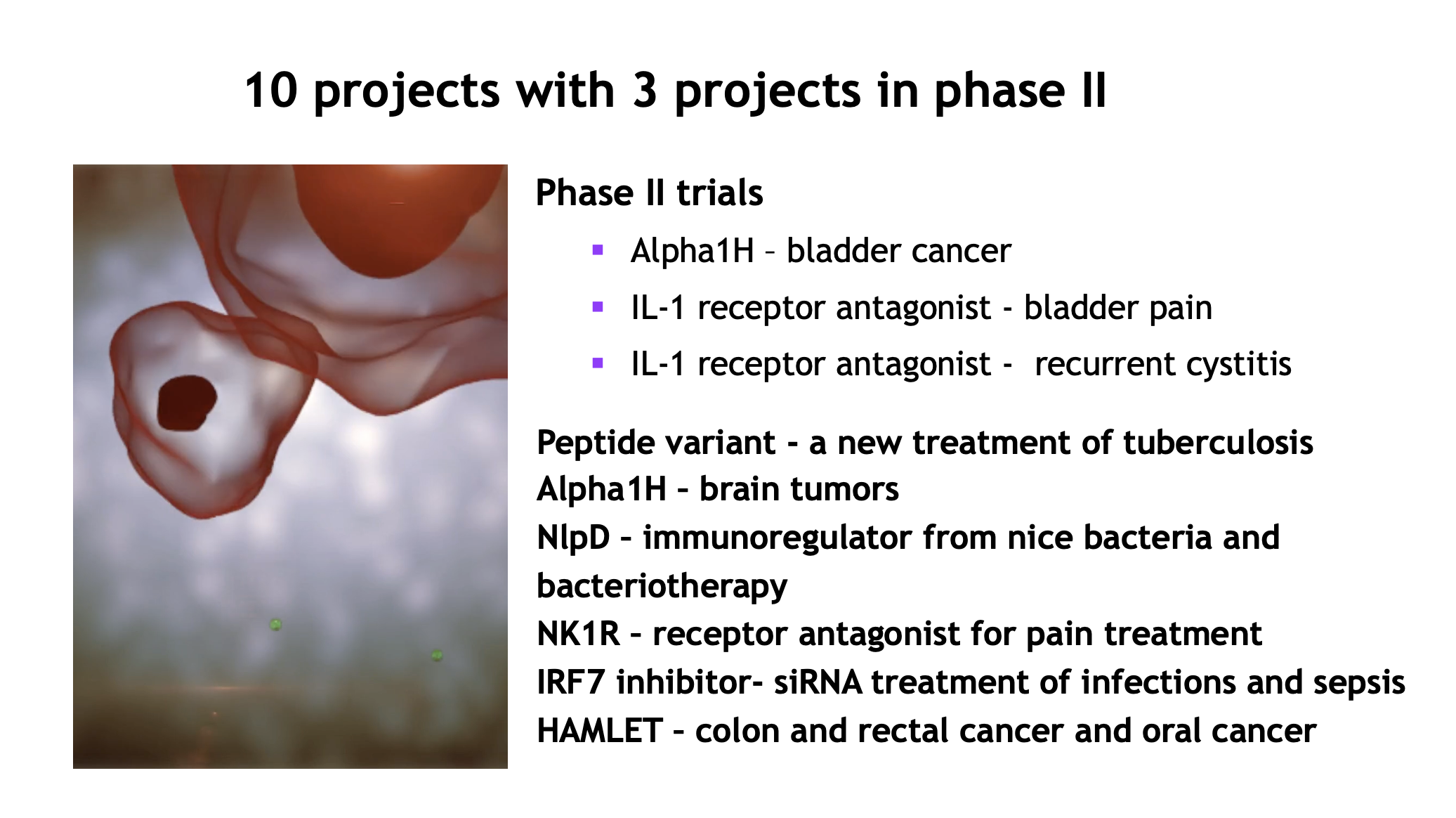
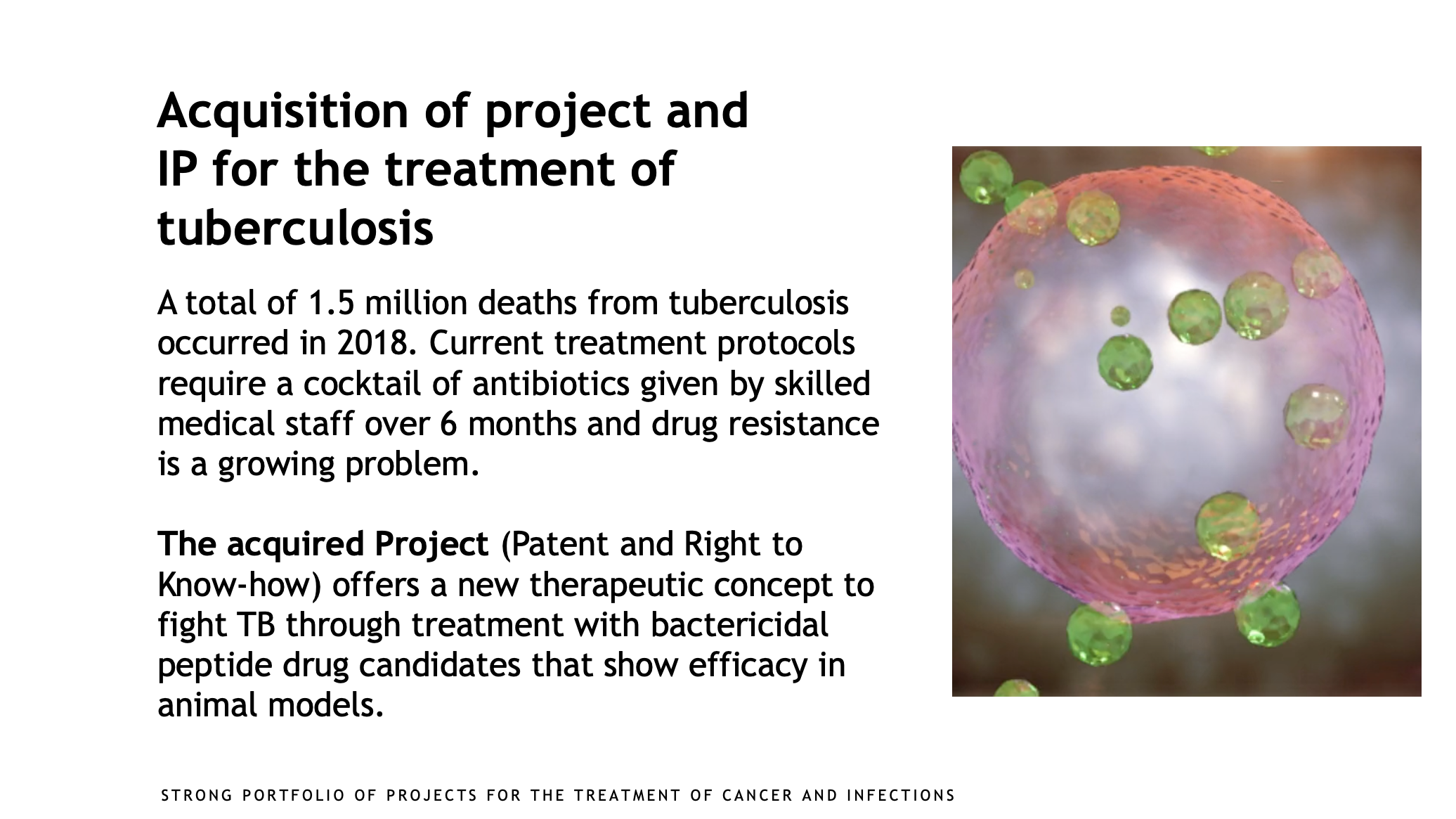
An innovative pharmaceutical company with a broad and strong portfolio of projects for the treatment of cancer and infections.










About Hamlet Biopharma




![]()
© 2024 Hamlet BioPharma.